This section provides information about the compilers at HPC2N. There are instructions about their usage, and some of the most popular flags, as well as the provided libraries and how to link to them.
There is also a section with an introduction to makefiles, a few examples, and links to more information.
Installed compilers¶
There are compilers available for Fortran 77, Fortran 90, Fortran 95, C, and C++. The compilers can produce both general-purpose code and architecture-specific optimized code to improve performance (loop-level optimizations, inter-procedural analysis and cache optimizations).
Compilers and Compiler Toolchains¶
The compilers are arranged in ‘Compiler toolchains’, which are bundles of software making up a complete environment for compiling or using a specific prebuilt software. A toolchain usually includes a compiler suite, an MPI version, BLAS, LAPACK, ScaLapack, and FFTW. It may also be just a compiler, like GCC alone, or something in between.
If you do
or
when you have logged in, you will get a list of (mainly) the compiler toolchains and their respective versions. You should always specify the full version when loading a compiler or software package.
Currently available toolchains:
- GCC: GCC only
- foss: GCC, OpenMPI, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- fosscuda: gompic, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- gcccuda: GCC and CUDA
- gomkl: GCC, OpenMPI, IntelMKL
- gompi: GCC, OpenMPI
- gompic: GCC, OpenMPI, CUDA
- iccifort: icc, ifort
- iccifortcuda: icc, ifort, CUDA
- iimpi: icc, ifort, IntelMPI
- imkl: icc, ifort, IntelMKL
- intel: icc, ifort, IntelMPI, IntelMKL
- intelcuda: intel, CUDA
- iompi: icc, ifort, OpenMPI
Use
to see which versions are available.
In order to be able to use compiler toolchain, you need to load it with
For more information about compiler toolchains, see our page about the module system.
Build environment¶
Using a compiler toolchain by itself is possible but requires a fair bit of manual work, figuring out which paths to add to -I or -L ifor including files and libraries, and similar.
To make life as a software builder easier there is a special module available, buildenv, that can be loaded on top of any toolchain. If it is missing for some toolchain, send a mail to support@hpc2n.umu.se and let us know.
This module defines a large number of environment variables with the relevant settings for the used toolchain. Among other things it sets CC, CXX, F90, FC, MPICC, MPICXX, MPIF90, CFLAGS, FFLAGS, and much more.
To see all of them, after loading a toolchain do:
Depending on the software one can use these environment variables to set related makefile variables or cmake defines, or just use them for guidelines on what to use in makefiles etc.
Exactly how to use them depends on the softwares build system.
Compiler usage¶
Compilers are preferrably loaded as part of compiler toolchains. Read more in the section Installed compilers and on our page about the module system for more about that.
OpenMP: All compilers has this included, so it is enough to load the module for a specific compiler toolchain and then add the appropriate flag (see Flags).
Note that instead of using the compiler names for the specific compiler (Intel, GCC) you can instead load the buildenv module after loading the compiler toolchain to get the environment variables that can be used in place of the compiler name.
GNU Compiler Collection (GCC)¶
The compiler toolchain modules that are using the GCC compilers:
- GCC: GCC only
- foss: GCC, OpenMPI, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- fosscuda: gompic, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- gcccuda: gcccuda: GCC and CUDA
- gomkl: GCC, OpenMPI, IntelMKL
- gompi: GCC, OpenMPI
- gompic: GCC, OpenMPI, CUDA
Remember to load a specific version (see available ones with ml av TOOLCHAIN-MODULE).
Compiling with GCC
| Language | Compiler name | MPI |
|---|---|---|
| Fortran77 | gfortran | mpif77 |
| Fortran90 | gfortran | mpif90 |
| Fortran95 | gfortran | N/A |
| C | gcc | mpicc |
| C++ | g++ | mpiCC |
There are more information to find on the man pages which can be found by typing:
Note that you need to load the compiler module first.
Here is a link to a place with more information: GCC online docs.
Intel Compilers¶
The Intel compilers are installed on Kebnekaise. The compiler toolchain modules that are using the Intel compilers:
- iccifort: icc, ifort
- iccifortcuda: icc, ifort, CUDA
- iimpi: icc, ifort, IntelMPI
- imkl: icc, ifort, IntelMKL
- intel: icc, ifort, IntelMPI, IntelMKL
- intelcuda: intel, CUDA
- iompi: icc, ifort, OpenMPI
Remember to load a specific version (see available ones with ml av TOOLCHAIN-MODULE).
Compiling with Intel
| Language | Compiler name | MPI |
|---|---|---|
| Fortran77 | ifort | mpiifort |
| Fortran90 | ifort | mpiifort |
| Fortran95 | ifort | N/A |
| C | icc | mpiicc |
| C++ | icpc | mpiicc |
There are more information to find on the man pages which can be found by typing:
Remember that you need to load the module first!
Here are some links to places with more information:
- Intel Fortran Compiler Classic and Intel Fortran Compiler Developer Guide and Reference
- Intel® C++ Compiler Classic Developer Guide and Reference
- Resource & Documentation Center
- Intel® oneAPI DPC++/C++ Compiler
- Developer Zone
Compiler flags¶
This page contains information about some of the more important/popular flags for the compilers available at HPC2N. The flags below can all be taken to be valid for the Fortran and C/C++ compilers alike, as well as for compiling with the MPI libraries included (remember to load the proper modules - see the page about the module system and the section about installed compilers for more information about that.)
Intel Compilers¶
The Intel compilers are installed on HPC2N’s systems.
List of commonly used flags:
- -fast This option maximizes speed across the entire program.
- -g Produce symbolic debug information in an object file. The compiler does not support the generation of debugging information in assemblable files. If you specify the -g option, the resulting object file will contain debugging information, but the assemblable file will not. The -g option changes the default optimization from -O2 to -O0. It is often a good idea to add -traceback also, so the compiler generates extra information in the object file to provide source file traceback information.
- -debug all Enables generation of enhanced debugging information. You need to also specify -g
- -O0 Disable optimizations. Use if you want to be certain of getting correct code. Otherwise use -O2 for speed.
- -O Same as -O2
- -O1 Optimize to favor code size and code locality. Disables loop unrolling.
- -O1 may improve performance for applications with very large code size, many branches, and execution time not dominated by code within loops. In most cases, -O2 is recommended over -O1.
- -O2 (default) Optimize for code speed. This is the generally recommended optimization level.
- -O3 Enable -O2 optimizations and in addition, enable more aggressive optimizations such as loop and memory access transformation, and prefetching. The -O3 option optimizes for maximum speed, but may not improve performance for some programs. The -O3 optimizations may slow down code in some cases compared to -O2 optimizations. Recommended for applications that have loops with heavy use of floating point calculations and process large data sets.
- -Os Enable speed optimizations, but disable some optimizations that increase code size for small speed benefit.
- -fpe{0,1,3} Allows some control over floating-point exception (divide by zero, overflow, invalid operation, underflow, denormalized number, positive infinity, negative infinity or a NaN) handling for the main program at runtime. Fortran only. Default is -fpe3 meaning all floating-point exceptions are disabled and floating-point underflow is gradual, unless you explicitly specify a compiler option that enables flush-to-zero. The default value may slow runtime performance.
- -qopenmp Enable the parallelizer to generate multi-threaded code based on the OpenMP directives. The code can be executed in parallel on both uniprocessor and multiprocessor systems.
- -parallel Enable the auto-parallelizer to generate multi-threaded code for loops that can be safely executed in parallel. The -parallel option enables the auto-parallelizer if either the -O2 or -O3 optimization option is also on (the default is -O2). You might need to set the
KMP_STACKSIZEenvironment variable to an appropriately large size, like 16m, to use this option.
To read about other flags, and for further information, look in the man files. They can be accessed like this:
Here are some links to places with more information:
- Intel Fortran Compiler Documentation
- Intel C++ Compiler Options
- Recommended Intel Compiler Debugging Options
GNU Compiler Collection (GCC)¶
List of commonly used flags:
- -o file Place output in file ‘file’.
- -c Compile or assemble the source files, but do not link.
- -fopenmp Enable handling of the OpenMP directives.
- -g Produce debugging information in the operating systems native format.
- -O or -O1 Optimize. The compiler tried to reduce code size and execution time.
- -O2 Optimize even more. GCC performs nearly all supported optimizations that do not involve a space-speed tradeoff.
- -O3 Optimize even more. The compiler will also do loop unrolling and function inlining. RECOMMENDED
- -O0 Do not optimize. This is the default.
- -Os Optimize for size.
- -Ofast Disregard strict standards compliance. -Ofast enables all -O3 optimizations. It also enables optimizations that are not valid for all standard-compliant programs. It turns on -ffast-math and the Fortran-specific -fno-protect-parens and -fstack-arrays.
- -ffast-math Sets the options -fno-math-errno, -funsafe-math-optimizations, -ffinite-math-only, -fno-rounding-math, -fno-signaling-nans and -fcx-limited-range.
- -l library Search the library named ‘library’ when linking.
To read about other flags, and for further information, look in the man files. They can be accessed by first loading the module (see section Installed Compilers and then doing either:
Here are links to places with more information:
Libraries¶
This page is about software libraries. The libraries on HPC2N systems includes parallel communications libraries (MPI) as well as various mathematical libraries, including MKL. More detailed documentation on the libraries available follows below.
To access the libraries you need to load a module. More specifically you load the libraries together with a compiler in a compiler toolchain (see the section ‘Installed compilers’).
Build environment¶
Using the libraries available through a compiler toolchain by itself is possible but requires a fair bit of manual work, figuring out which paths to add to -I or -L for include files and libraries, and similar.
To make life as a software builder easier there is a special module available, buildenv, that can be loaded on top of any toolchain. If it is missing for some toolchain, send a mail to support@hpc2n.umu.se and let us know.
This module defines a large number of environment variables with the relevant settings for the used toolchain. Among other things it sets CC, CXX, F90, FC, MPICC, MPICXX, MPIF90, CFLAGS, FFLAGS, and much more.
To see all of them, load a toolchain and do
Depending on the software one can use these environment variables to set related makefile variables or cmake defines, or just use them for guidelines on what to use in makefiles etc.
Exactly how to use them depends on the softwares build system.
You will now get a list resembling this (I have colourized some of the common libraries).
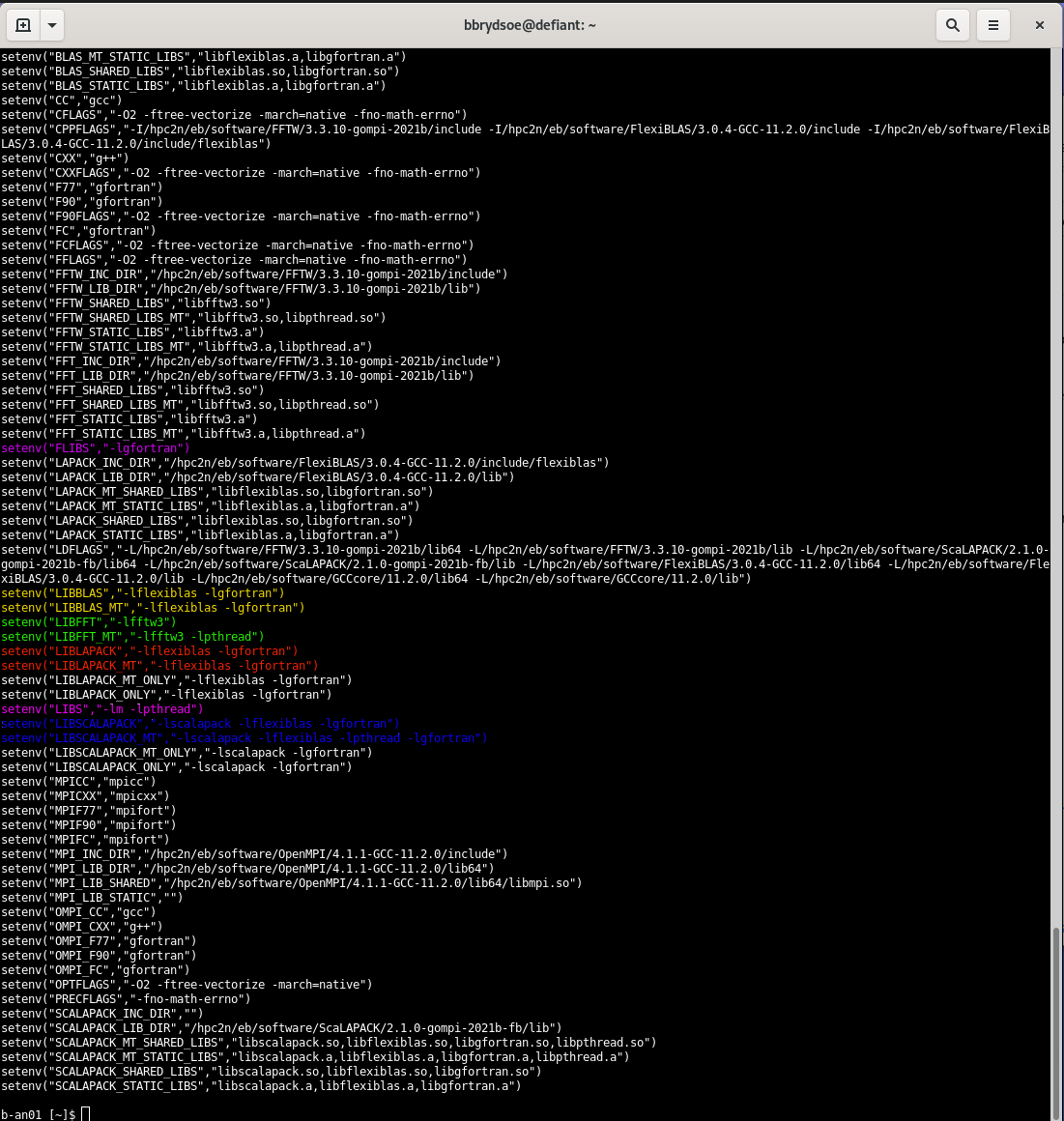{kind=link}
There are some variables that ends in “_MT”, they should be used if threaded versions of the libraries are needed.
Note: It is highly recommended to use the enviroment variables from the buildenv module.
MPI Libraries¶
Message Passing Interface (MPI) is a standardized and portable message-passing system designed to function on a wide variety of parallel computing architectures. Several implementations exist, among others OpenMPI and Intel MPI.
A number of compiler toolchains at HPC2N has OpenMPI and Intel MPI installed. They are best loaded using one of the following compiler toolchains:
- foss: GCC, OpenMPI, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- fosscuda: gompic, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- gomkl: GCC, OpenMPI, IntelMKL
- gompi: GCC, OpenMPI
- gompic: GCC, OpenMPI, CUDA
- iimpi: icc, ifort, IntelMPI
- imkl: icc, ifort, IntelMKL
- intel: icc, ifort, IntelMPI, IntelMKL
- intelcuda: intel, CUDA
- iompi: icc, ifort, OpenMPI
To compile something, first load the compiler toolchain module with:
and then use the appropriate mpi wrapper command:
| Language | Command, gcc | Command, intel |
|---|---|---|
| Fortran 77 | mpif77 | mpiifort |
| Fortran 90 | mpif90 | mpiifort |
| Fortran 95 | mpif90 | mpiifort |
| C | mpicc | mpiicc |
| C++ | mpicxx | mpiicpc |
To run, you need to add this to your job submit script:
Here are a few links to pages with more information about the different implementations of the MPI libraries:
- OpenMPI, external documentation: https://www.open-mpi.org/
- Intel MPI, external documentation: Intel MPI Library
Math (and other) Libraries¶
The following list is not exhaustive, but it covers the most ‘popular’ of the libraries that are installed at HPC2N. Note that Python libraries are not included here.
- BLAS
- BLACS
- Eigen
- ELPA
- FFTW
- GSL
- Intel MKL
- LAPACK
- Libint
- libxc
- Libxsmm
- METIS
- MPFR
- NetCDF
- ParMETIS
- ScaLAPACK
- SCOTCH
- SIONlib
- StarPU
Use:
to see a more complete list of modules, including libraries.
In some cases the libraries are loaded as separate modules, but in most cases you need to first load a suitable compiler toolchain.
You can also use Intel MKL, recommended for compiling with the Intel compilers.
Examples of linking with math libraries
NOTE: in all the examples I use -o PROGRAM to name the executable PROGRAM. If you leave out this your executable will be named a.out.
BLAS¶
Blas is available in the form of FlexiBLAS, OpenBLAS, or Intel MKL. Intel MKL is often recommended if you are compiling with Intel compilers. See the section about Intel MKL for more information about that.
Linking with FlexiBLAS:
FlexiBLAS - A BLAS and LAPACK wrapper library with runtime exchangable backends.
You can load it as a module after first loading a suitable GCC module (ml spider FlexiBLAS for more information). It is also available as part of several compiler toolchain versions, like foss/2021a and the newer foss versions. Remember, you can always see which other modules are included in a toolchain with ml show TOOLCHAIN/VERSION.
| Language | Command |
|---|---|
| Fortran 77 | gfortran -o PROGRAM PROGRAM.f -lflexiblas -lgfortran |
| Fortran 90 | gfortran -o PROGRAM PROGRAM.f90 -lflexiblas -lgfortran |
| C | gcc -o PROGRAM PROGRAM.c -lflexiblas -lgfortran |
| C++ | g++ -o PROGRAM PROGRAM.cc -lflexiblas -lgfortran |
Or use the environment variable to link with: $LIBBLAS.
Linking with OpenBLAS:
OpenBLAS is an optimized BLAS library based on GotoBLAS2 1.13 BSD version.
Load it either as a module after first loading a suitable GCC module (ml spider OpenBLAS for more information), or as part of several compiler toolchain versions. You can find it in foss/2020b and older versions of foss, as well as fosscuda/2020b and older. Remember, you can always see which other modules are included in a toolchain with ml show TOOLCHAIN/VERSION.
| Language | Command |
|---|---|
| Fortran 77 | gfortran -o PROGRAM PROGRAM.f -lopenblas -lgfortran |
| Fortran 90 | gfortran -o PROGRAM PROGRAM.f90 -lopenblas -lgfortran |
| C | gcc -o PROGRAM PROGRAM.c -lopenblas -lgfortran |
| C++ | g++ -o PROGRAM PROGRAM.cc -lopenblas -lgfortran |
Or use the environment variable to link with: $LIBBLAS. This requires you to load the buildenv module after loading the compiler toolchain.
LAPACK¶
LAPACK is written in Fortran77 and provides routines for solving systems of simultaneous linear equations, least-squares solutions of linear systems of equations, eigenvalue problems, and singular value problems. The associated matrix factorizations (LU, Cholesky, QR, SVD, Schur, generalized Schur) are also provided, as are related computations such as reordering of the Schur factorizations and estimating condition numbers. Dense and banded matrices are handled, but not general sparse matrices. In all areas, similar functionality is provided for real and complex matrices, in both single and double precision.
Link to external information about LAPACK.
You can also use the Intel MKL version of LAPACK.
Linking with LAPACK and BLAS:
The Fortran based lapack library is included with the BLAS modules. To use it you must load the BLAS module you want, as well as its prerequisite compiler (toolchain).
Load a suitable version of either foss, fosscuda, or a GCC compiler suite.
Do ml av to see which versions you can load. Currently, foss/2020b and older versions of foss, as well as fosscuda/2020b and older are OpenBLAS, the newer ones are FlexiBLAS.
Use the following commands to compile and link.
OpenBLAS
| Language | Command |
|---|---|
| Fortran 77 | gfortran -o PROGRAM PROGRAM.f -lopenblas -lgfortran |
| Fortran 90 | gfortran -o PROGRAM PROGRAM.f90 -lopenblas -lgfortran |
| C | gcc -o PROGRAM PROGRAM.c -lopenblas -lgfortran |
| C++ | g++ -o PROGRAM PROGRAM.cc -lopenblas -lgfortran |
FlexiBLAS
| Language | Command |
|---|---|
| Fortran 77 | gfortran -o PROGRAM PROGRAM.f -lflexiblas -lgfortran |
| Fortran 90 | gfortran -o PROGRAM PROGRAM.f90 -lflexiblas -lgfortran |
| C | gcc -o PROGRAM PROGRAM.c -lflexiblas -lgfortran |
| C++ | g++ -o PROGRAM PROGRAM.cc -lflexiblas -lgfortran |
Or use the environment variable $LIBLAPACK to link with. This requires you to load the buildenv module after loading the compiler toolchain.
BLACS¶
As of ScaLAPACK version 2, BLACS is now included in the ScaLAPACK library. (Link to external information about BLACS.)
ScaLAPACK¶
Since the usage of ScaLAPACK depends on LAPACK, it involves multiple libraries.
NOTE: As of version 2, ScaLAPACK includes BLACS. This means that it is tightly coupled to the MPI implementation used to build it. In order to use this library, a compiler and the corresponding MPI libraries needs to be loaded first, as well as ScaLAPACK, LAPACK and BLAS for that compiler. This is easily accomplished by loading a suitable compiler toolchain module.
Linking with ScaLAPACK, BLAS, and LAPACK
You can load either the foss or the fosscuda toolchain. Do ml av to see which versions you can load (you should always load a specific version). Currently, foss/2020b and older versions of foss, as well as fosscuda/2020b and older are OpenBLAS, the newer ones are FlexiBLAS.
In addition, you can use Intel MKL if you are using the Intel compilers.
After loading the compiler toolchain module, use the following command to compile and link with ScaLAPACK
Toolchain versions with OpenBLAS
| Language | Command |
|---|---|
| Fortran 77 | mpifort -o PROGRAM PROGRAM.f -lscalapack -lopenblas -lgfortran |
| Fortran 90 | mpifort -o PROGRAM PROGRAM.f90 -lscalapack -lopenblas -lgfortran |
| C | mpicc -o PROGRAM PROGRAM.c -lscalapack -lopenblas -lgfortran |
| C++ | mpicc -o PROGRAM PROGRAM.cc -lscalapack -lopenblas -lgfortran |
Toolchain versions with FlexiBLAS
| Language | Command |
|---|---|
| Fortran 77 | mpifort -o PROGRAM PROGRAM.f -lscalapack -lflexiblas -lgfortran |
| Fortran 90 | mpifort -o PROGRAM PROGRAM.f90 -lscalapack -lflexiblas -lgfor |
| tran | |
| C | mpicc -o PROGRAM PROGRAM.c -lscalapack -lflexiblas -lgfortran |
| C++ | mpicc -o PROGRAM PROGRAM.cc -lscalapack -lflexiblas -lgfortran |
Or use the environment variable, $LIBSCALAPACK to link with. This requires you to load the buildenv module after loading the compiler toolchain.
FFTW¶
The available versions of FFTW are all 3.x. Most have MPI support. See ml spider FFTW to see which are serial and which are MPI.
Link to external information about FFTW.
To use FFTW, you should load it as part of a compiler toolchain. The available modules are foss and fosscuda. Do ml av to see which versions you can load. In addition, you can use Intel MKL if you are using the Intel compilers.
Use these commands to compile and link with FFTW3
| Language | Command |
|---|---|
| Fortran 77 | gfortran -o PROGRAM PROGRAM.f -lfftw3 -lm |
| Fortran 90 | gfortran -o PROGRAM PROGRAM.f90 -lfftw3 -lm |
| C | gcc -o PROGRAM PROGRAM.c -lfftw3 -lm |
| C++ | g++ -o PROGRAM PROGRAM.cc -lfftw3 -lm |
Or use \(LIBFFT -lm</code> to link with (<code>\)LIBFFT_MT -lm for threaded). This requires you to load the buildenv module after loading the compiler toolchain.
METIS¶
METIS is a set of serial programs for partitioning graphs, partitioning finite element meshes, and producing fill reducing orderings for sparse matrices.
Link to external information about METIS.
To see which versions of METIS are available use:
Remember to load the prerequisite compiler suite or toolchain.
Use these commands to compile and link with METIS
| Language | Command |
|---|---|
| Fortran 77 | gfortran -o PROGRAM PROGRAM.f -lmetis |
| Fortran 90 | gfortran -o PROGRAM PROGRAM.f90 -lmetis |
| C | gcc -o PROGRAM PROGRAM.c -lmetis |
| C++ | g++ -o PROGRAM PROGRAM.cc -lmetis |
ELPA¶
The publicly available ELPA library provides highly efficient and highly scalable direct eigensolvers for symmetric matrices. Though especially designed for use for PetaFlop/s applications solving large problem sizes on massively parallel supercomputers, ELPA eigensolvers have proven to be also very efficient for smaller matrices.
Link to external information about ELPA.
To see which versions of ELPA are available use:
Remember to load any prerequisites before loading the ELPA module. Use ml spider ELPA/VERSION for each version of ELPA to see the prerequisites.
You can find the libraries that can be linked with in $EBROOTELPA/lib when the module has been loaded. In addition, there is a USERS_GUIDE.md file with information about how to use ELPA. It can be found in $EBROOTELPA/share/doc/elpa.
Eigen¶
Eigen is a C++ template library for linear algebra: matrices, vectors, numerical solvers, and related algorithms.
Link to external information about Eigen.
To see which versions of Eigen are available use:
Remember to load the needed prerequisites for the version (listed when you do ml spider Eigen/VERSION for the version you want).
You can find the Eigen library files under the $EBROOTEIGEN/lib directory after the module has been loaded.
There is a getting started guide and other documentation on the Eigen homepage.
GSL¶
The GNU Scientific Library (GSL) is a numerical library for C and C++ programmers. It is free software under the GNU General Public License.
Link to external information about GSL.
To see which versions of GSL are available use:
Then do
for the VERSION you would like to load, in order to see the prerequisites that are needed ti load the GSL module.
The GSL libraries can be found in $EBROOTGSL/lib after the module has been loaded, if you need to update yourself on their names.
After loading, you can get some information about GSL from the command
SCOTCH¶
Software package and libraries for sequential and parallel graph partitioning, static mapping, and sparse matrix block ordering, and sequential mesh and hypergraph partitioning.
Link to external information about SCOTCH.
To see which versions of SCOTCH are available, and how to load it and its dependencies, use:
Then check with ml spider for a specific version how it is loaded (which prerequisites needs to be loaded first).
When the module has been loaded, you can use the environment variable $EBROOTSCOTCH to find the binaries and libraries for SCOTCH.
The text files for the user manual on how to use SCOTCH can be found in $EBROOTSCOTCH/man/man1/ when the module is loaded. You can also copy the content of that directory and then run “make” to generate a full user manual.
Libint¶
Libint library is used to evaluate the traditional (electron repulsion) and certain novel two-body matrix elements (integrals) over Cartesian Gaussian functions used in modern atomic and molecular theory.
Link to external information about Libint.
To see which versions of Libint are available, and how to load it and any dependencies, use:
and then
for the specfic VERSION you are interested in.
When the module and its prerequisites have been loaded, you can use the environment variable $EBROOTLIBINT to find the binaries and libraries for Libint.
There is some information about Libint and how to use it on the Libint Homepage. There is a brief Libint Programmers Manual here.
Libxc¶
Libxc is a library of exchange-correlation functionals for density-functional theory. The aim is to provide a portable, well tested and reliable set of exchange and correlation functionals.
Link to external information about Libxc.
To see which versions of Libxc are available, and how to load it and any dependencies, use:
and then use ml spider to check a specific version to see how to load it and the prerequisites.
When the module has been loaded, you can use the environment variable $EBROOTLIBXC to find the binaries and libraries for Libxc.
There is a Libxc manual here.
Libxsmm¶
LIBXSMM is a library for small dense and small sparse matrix-matrix multiplications targeting Intel Architecture (x86).
Link to external information about Libxsmm.
To see which versions of Libxsmm are available, and how to load it and any dependencies, use:
and then use ml spider to check a specific version to see how to load it.
When the module has been loaded, you can use the environment variable $EBROOTLIBXSMM to find the binaries and libraries for Libxsmm.
There is some Libxsmm documentation here.
MPFR¶
The MPFR library is a C library for multiple-precision floating-point computations with correct rounding.
Link to external information about MPFR.
To see which versions of MPFR are available, and how to load it and any dependencies, use:
and then do ml spider on a specific version to see how to load it.
When the module has been loaded, you can use the environment variable $EBROOTMPFR to find the binaries and libraries for MPFR.
The MPFR Reference Guide is here (for the newest version).
NetCDF¶
NetCDF (Network Common Data Form) is a set of software libraries and self-describing, machine-independent data formats that support the creation, access, and sharing of array-oriented scientific data.
Link to external information about NetCDF.
To see which versions of NetCDF are available, and how to load it and any dependencies, use:
and then do ml spider on a specific version to see how to load it.
When the module has been loaded, you can use the environment variable $EBROOTNETCDF to find the binaries and libraries for NetCDF.
There is some information about NetCDF and how to use it on the NetCDF documentation page.
There is also a Parallel netCDF available, for parallel I/O access. The module is called PnetCDF.
ParMETIS¶
ParMETIS is an MPI-based parallel library that implements a variety of algorithms for partitioning unstructured graphs, meshes, and for computing fill-reducing orderings of sparse matrices.
Link to external information about ParMETIS.
To see which versions of ParMETIS are available, and how to load it and any dependencies, use:
and then use ml spider on a specific version to see how to load it.
When the module has been loaded, you can use the environment variable $EBROOTPARMETIS to find the binaries and libraries for ParMETIS.
There is a brief ParMETIS manual here.
SIONlib¶
SIONlib is a scalable I/O library for parallel access to task-local files. The library not only supports writing and reading binary data to or from several thousands of processors into a single or a small number of physical files, but also provides global open and close functions to access SIONlib files in parallel. This package provides a stripped-down installation of SIONlib for use with performance tools (e.g., Score-P), with renamed symbols to avoid conflicts when an application using SIONlib itself is linked against a tool requiring a different SIONlib version.
Link to external information about SIONlib.
To see which versions of SIONlib are available, and how to load it and any dependencies, use:
and then do ml spider on a specific version to see how to load it.
When the module has been loaded, you can use the environment variable $EBROOTSIONLIB to find the binaries and libraries for SIONlib.
There is some documentation for SIONlib here.
CUDA libraries¶
NOTE: CUDA libraries can be used with either GCC or Intel compilers. In addition, the NVIDIA CUDA compiler driver nvcc is installed.
You should load one of the following compiler toolchain modules:
- fosscuda: gompic, OpenBLAS/LAPACK, FFTW, ScaLAPACK
- gcccuda: GCC and CUDA
- gompic: GCC, OpenMPI, CUDA
- iccifortcuda: icc, ifort, CUDA
- intelcuda: intel, CUDA
in a suitable version (check with ml spider for the compiler toolchain).
After you have loaded the compiler toolchain module, you compile and link with CUDA like this:
| Language | GCC, OpenMPI | Intel, Intel MPI | NVCC |
|---|---|---|---|
| Fortran calling CUDA functions | 1) nvcc -c CUDAPROGRAM.cu 2) gfortran -lcudart -lcuda PROGRAM.f90 CUDAPROGRAM.o |
||
| C / C++ with CUDA | mpicc CUDAPROGRAM.cu -lcuda -lcudart | mpiicc CUDAPROGRAM.cu -lcuda -lcudart | nvcc CUDAPROGRAM.cu |
You can add other flags, like for instance -o my-binary to name the output differently than the standard a.out.
NOTE: CUDA functions can be called directly from Fortran programs:
- First use the nvcc compiler to create an object file from the .cu file.
- Then compile the Fortran code together with the object file from the .cu file.
Intel MKL libraries¶
The Intel MKL libraries contains:
- ScaLAPACK
- LAPACK
- Sparse Solver
- BLAS
- Sparse BLAS
- PBLAS
- GMP
- FFTs
- BLACS
- VSL
- VML
More information about MKL and the libraries in it can be found at the external links:
Linking with MKL libraries
To use the MKL libraries load one of the following compiler toolchain modules:
- gomkl: GCC, OpenMPI, IntelMKL
- imkl: icc, ifort, IntelMKL
- intel: icc, ifort, IntelMPI, IntelMKL
- intelcuda: intel, CUDA
in a suitable version (check with ml spider for the relevant compiler toolchain).
To correctly use MKL it is vital to have read the documentation. To find the correct way of linking, take a look at the offical Intel MKL documentation.
Using the buildenv module, the common blas/lapack/scalapack/fftw libraries are available in the following environment variables, just like when using a non-MKL capable toolchain:
- LIBBLAS
- LIBLAPACK
- LIBSCALAPACK
- LIBFFT
And threaded versions are available from the corresponding environment variable appended with “_MT”
Read the section about buildenv for more information.
There are too many libraries in MKL to show a complete list of combinations. We refer you to the official MKL documentation for examples and support@hpc2n.umu.se for help.
makefiles¶
The purpose of makefiles is to help you with your coding projects. One will often have a program which have, say, several header files and a number of C source files. If you then make a change to one of the files, you will have to recompile all the files. This is handled by the program make, following the commands in your makefile - it will only recompile the files which contains changes, and also makes it easier and faster to give the order to compile everything. You no longer have to write long and complex compiler commands every time.
Usually, the file that contains all the commands to make is called makefile or Makefile. You can then just invoke make from the command line and it will follow these instructions. You can, however, give the file any name. If you call it something else you will have to use make -f NAME_OF_MAKEFILE to call it.
Structure¶
A makefile consists of three sections: target, dependencies, and rules. The target is normally either an executable or object file name. The dependencies are source code or other things needed to make the target. The rules are the commands needed to make the target.
A simple makefile might be structured like this:
A makefile can also contain definitions of variables and inclusion of other makefiles. The variables in makefiles may be overridden in the command-line arguments that are passed to the make utility. An example is that the variable “CC” is often used in makefiles to refer to a specific C compiler, and the user may wish to provide an alternate compiler to use.
NOTE: A very important thing to remember about makefiles are that every rule line begins with a tab, not spaces. Otherwise make might think it is some sort of dependency.
Comments¶
Any line that begins with a # is a comment and will be ignored.
Explicit rules¶
These are rules that tell make which files depend on the compilation of other files, as well as the commands required to compile a particular file. They have this form:
This rule means that to create the targetfile, make must perform the listed commands on the sourcefiles.
Example
The above example shows a rule that says that in order to create the targetfile main, the sourcefiles main.c, test.h and List.h have to exist, and make should use the command:
to create it.
Implicit rules¶
Implicit rules are in many ways like explicit rules, they are just listed without commands. This means make will use the suffixes on the files to determine which commands to perform.
Example
This will cause the following command to be executed:
Variables¶
It is possible to define variables in a makefile. To do this use the following command:
It is a convention to write variable names in uppercase. For instance:
In order to get a variable’s value, put the symbol $ before the varable’s name, like this:
Automatic Variables¶
(Much of this section is taken from https://www.gnu.org/software/make/manual/html_node/Automatic-Variables.html#Automatic-Variables).
Suppose you are writing a pattern rule to compile a “.c” file into an “.o” file: how do you write the “cc” command so that it operates on the right source file name? You cannot write the name in the recipe, because the name is different each time the implicit rule is applied.
What you do is use another feature of make, namely automatic variables, which are computed afresh for each rule that is executed, based on the target and prerequisites of the rule. For example, you would use “$@” for the object file name and “$<” for the source file name.
NOTE that automatic variables:
- only have values within the recipe - you cannot use them anywhere within the target list of a rule (they have no value there and will expand to the empty string).
- they cannot be accessed directly within the prerequisite list of a rule. A common mistake is attempting to use
$@within the prerequisites list; this will not work.
| Automatic variables | |
|---|---|
| $@ | The file name of the target of the rule. If the target is an archive member, then “$@” is the name of the archive file. In a pattern rule that has multiple targets, “$@” is the name of whichever target caused the rule’s recipe to be run. |
| $% | The target member name, when the target is an archive member. For example, if the target is foo.a(bar.o) then “$%” is bar.o and “$@” is foo.a. “$%” is empty when the target is not an archive member. |
| $< | The name of the first prerequisite. If the target got its recipe from an implicit rule, this will be the first prerequisite added by the implicit rule. |
| $? | The names of all the prerequisites that are newer than the target, with spaces between them. For prerequisites which are archive members, only the named member is used |
| $^ | The names of all the prerequisites, with spaces between them. For prerequisites which are archive members, only the named member is used. A target has only one prerequisite on each other file it depends on, no matter how many times each file is listed as a prerequisite. So if you list a prerequisite more than once for a target, the value of “$^” contains just one copy of the name. This list does not contain any of the order-only prerequisites; for those see the “$|” variable, below. |
| $+ | This is like “$^“, but prerequisites listed more than once are duplicated in the order they were listed in the makefile. This is primarily useful for use in linking commands where it is meaningful to repeat library file names in a particular order. |
| $| | The names of all the order-only prerequisites, with spaces between them. |
| $* | The stem with which an implicit rule matches. If the target is dir/a.foo.b and the target pattern is a.%.b then the stem is dir/foo. The stem is useful for constructing names of related files. In a static pattern rule, the stem is part of the file name that matched the “ %” in the target pattern. In an explicit rule, there is no stem; so “ $*” cannot be determined in that way. Instead, if the target name ends with a recognized suffix, “$*” is set to the target name minus the suffix. For example, if the target name is “foo.c“, then “$*” is set to “foo“, since “.c” is a suffix. GNU make does this bizarre thing only for compatibility with other implementations of make. You should generally avoid using “$*” except in implicit rules or static pattern rules. |
Example:
Here:
$@evaluates to all$<evaluates tofile.c$^evaluates tofile.c main.c
Examples¶
This small makefile is started with either make or make Hello. It does one more thing than just compile the program - there is also the target “clean” which is used to remove all the files generated by the compilation with one command: make clean.
The first line in this makefile is the dependendencies (Hello: Hello.o), while the second line contains the rule to create the target Hello. The third line the dependencies for making Hello.o are listed, while line 4 contains the rules to make the target Hello.o in line 3.
Note that the PHONY tag is a technicality that tells make that a particular target name does not produce an actual file. It is not strictly necessary to include the line with the PHONY tag, unless a file named clean exists in the current directory. The “$@” and “$\<” are two of the so-called internal macros and stand for the target name and “implicit” source, respectively. There are a number of other internal macros, which can be seen here.
main.exe : main.o foo.o
gcc main.o foo.o -o main.exe
main.o : main.c
gcc -c main.c
foo.o : foo.c
gcc -c foo.c
This simple makefile is run by giving the command make. The program always starts with the first rule it sees, in this case for main.exe. It then tries to build that first and finds dependencies. It will then check to see if any of those needs to be updated. This means, that the first time you call make for this makefile, it will run the following three commands:
Let us assume you then edit the file foo.c and run make again. It will then only run these commands:
Because there was no reason to recompile main.c, it will not do that.
makefile with variables
There are many similarities between this makefile and the last. The difference is that many of the commands have been replaced with variables. Then, when make runs, it replaces the variables in the target, dependency, and command sections of the rules. This is often an advantage, since it lets you specify the things that changes often in one place. One example is that it is very easy to change the compiler, since we are using the variable CC everywhere - and it is defined at the very top.
makefile with patterns
This is almost the same makefile as the one above, however, if you look in the last line you will see a pattern has been defined. This can be used every time make needs to compile any source. This is much simpler if we have many sources - otherwise we would have to write a rule for including the source for each of them. In the pattern, the “%” character is used to denote the part of the target and dependency that matches whatever the pattern is used for. The “$<” is a special variable meaning “whatever the dependencies are”. There is another useful variable which we will often have use for. It is “$@“, which means “the target”.
After this addition we can now add more source files in a simple manner, by just updating the line that defines the OBJECTS variable. The variable names CC and CFLAGS are part of make’s built-in rules. To see a list of all of these, look at man make or run make -p. There is also a reference manual for make, which contains many examples and suggestions for building makefiles.
Links¶
Here is a list of links to more information about makefiles.